青岛科技大学Adv Mat丨亚纳米铁纳米簇:解锁法拉第电容去离子中的超快动力学、卓越稳定性及明确机理
- 2026-07-18 04:14:01

超小金属纳米簇(MNCs,<2 nm)是一类具有独特性能的新兴材料,但非贵金属纳米簇(尤其是铁、钛、锰等高熔点元素)的合成仍面临挑战,缺乏温和、通用的制备策略,限制了其在电化学脱盐等领域的应用。本文报道了一种通用的 “孔介导气相扩散”(PVD)方法,在介孔碳球中合成出亚纳米铁纳米簇(0.8 nm),解决了法拉第电容去离子(FDI)这一缓解全球水资源短缺的关键技术瓶颈。该方法无需高温条件,且可拓展至其他难熔金属。作为 FDI 阳极,铁纳米簇电极实现了 116.83 mg₍Cl₎・g⁻¹ 的创纪录盐吸附容量、0.57 mg₍Cl₎・g⁻¹・s⁻¹ 的超高速率以及卓越的稳定性(200 次循环后容量保持率 86.47%)。亚纳米结构赋予材料超快离子扩散能力和应力缓解特性,突破了 FDI 中长期存在的动力学和稳定性限制。通过原位X射线光谱与密度泛函理论(DFT)计算,明确氯离子储存机理为转化反应(Fe 纳米簇 + Cl⁻ ⇔ FeOCl),在原子层面解决了铁基电化学中的关键机理争议。该研究为非贵金属纳米簇提供了通用合成平台,同时提供了原子级别的机理认知,推动下一代脱盐技术的发展。
科学问题
金属纳米簇(MNCs)指核心尺寸小于 2 nm 的材料,是一类新兴功能材料,具有原子级精确结构、可调表面化学性质、高比表面积以及类分子特性(如光致发光、手性)等独特优势。这些特性使其在催化、能量转换/存储、疾病诊疗和环境保护等领域引发广泛研究兴趣。然而,尽管铋、镍、锰、铁等非贵金属纳米簇在成本、资源可及性、本征物理化学性质及应用范围上具有显著优势,其发展却明显滞后于贵金属纳米簇。这一差距主要源于缺乏适用于非贵金属的合成策略 —— 传统贵金属纳米簇的制备方法无法直接套用。模板浸渍等常规方法制备的材料质量差、尺寸分布宽(从单原子到纳米颗粒),难以满足需求。近年来,多孔基质中金属颗粒的温度介导汽化(自上而下法)等技术虽实现了铋、锑等低熔点金属的亚纳米簇合成,但存在关键局限性:i)热稳定性限制使其无法应用于铁、钛、锰等高熔点金属;ii)极端温度(>800℃)导致反应动力学失控、尺寸均一性差且能耗高;iii)合成效率本质上较低。因此,开发通用、温和、高效的高质量非贵金属纳米簇合成策略至关重要,不仅能完善纳米簇材料体系,还能解锁其在能量存储、电化学脱盐和水消毒等关键领域应对挑战的潜力。
法拉第电容去离子(FDI)是一种极具前景的电化学脱盐技术,其核心通过电极氧化还原反应实现离子储存,具有脱盐性能优异、充电效率高、水资源利用率高等优势。尽管器件结构方面已取得进展,但电极材料带来的固有挑战 —— 脱盐速率低和循环稳定性差 —— 仍未解决。对铋、银、铁基材料等常见阳极的分析表明,其体相特性和低表体原子比导致阴离子储存动力学迟缓,或体相结合过程中体积膨胀严重。将电极材料尺寸缩小至纳米簇尺度(<2 nm)是合理的解决方案:纳米簇的超小尺寸提供极高的表面原子比例,大幅缩短离子扩散路径,显著提升脱盐动力学;同时,其固有的结构灵活性赋予材料优异的体积变化耐受性,改善循环稳定性。铁基纳米材料因氯离子储存容量高、成本低、毒性小,成为极具潜力的FDI阳极。但铁的高熔点(1538℃)使其电极形式长期局限于体相晶体结构(如 β-FeOOH),亚纳米铁纳米簇的合成与应用成为一项艰巨但关键的挑战。
此外,体相电极还带来一个根本性问题:FDI 中氯离子储存的原子级机理尚不明确。传统体相体系因反应导向的固有结构异质性,难以实现明确的原子级表征,导致这一知识缺口难以弥补。因此,现有研究仅提出现象学模型(如氯离子在体相β-FeOOH 晶体通道中的嵌入),缺乏原子分辨率的理解,严重限制了研究进展。尽管经过多年研究,核心问题仍未解决 —— 反应产物究竟是 FeCl₃还是 FeOCl,这已成为 FDI 合理设计的重大瓶颈。亚纳米铁纳米簇为解决这一问题提供了变革性方案:除解决动力学和稳定性限制外,其原子级均一性和明确结构便于开展先进表征,能够直接观察并明确阐释原子级别的氯离子储存机理。这种从体相设计到纳米簇工程的范式转变,为下一代FDI阳极奠定基础,并建立了通过亚纳米反应控制实现高效脱盐的通用方法。
概念验证
本研究通过两步 PVD 策略在介孔空心碳球(HCS)中合成亚纳米铁纳米簇(图 1a)。首先,采用传统硬模板法制备具有均匀介孔结构和明确中空腔的 HCS;其次,以乙酰丙酮铁(Fe (acac)₃)为铁前驱体,与 HCS 充分混合后密封在安瓿瓶中(真空度≈10⁻⁵ mbar),采用双温调制程序:先加热至 160℃保持 48 h,使前驱体升华(真空下升华温度 70-90℃),生成气态乙酰丙酮铁分子,均匀扩散到 HCS 介孔中并通过孔道缺陷有效锚定;随后升温至 240℃保持 24 h,使锚定的前驱体分子热解(热解起始温度 > 160-200℃),在孔内形成铁纳米簇;最后将安瓿瓶在水中快速淬火,得到HCS负载的铁纳米簇(Fe NCs@HCS)。该方法的巧妙之处在于利用乙酰丙酮铁升华和分解的不同温度窗口,实现碳介孔内分子级均匀分散,达成铁纳米簇的原子级精确合成(图 1b-h)。为进行后续 FDI 对比实验,通过增加乙酰丙酮铁负载量,在相同条件下制备了较大尺寸的铁纳米颗粒(Fe NPs@HCS)(图 1i-l)。
采用场发射扫描电子显微镜(FESEM)、透射电子显微镜(TEM)和高角环形暗场扫描透射电子显微镜(HAADF-STEM)对 HCS、Fe NCs@HCS 和 Fe NPs@HCS 的纳米结构进行表征。FESEM 和 TEM 结果(图 1c,j)显示,原始 HCS、Fe NCs@HCS 和 Fe NPs@HCS 均呈现均匀的球形结构(直径≈400 nm),且每个球形结构均具有明确的中心空腔,形成壳厚≈50 nm 的中空球形结构(图 S1d)。此外,所有样品的 HCS 壳层结构均含有径向排列的孔道(图 S2),可作为锚定位点和空间限制模板捕获气态铁前驱体,最终促进铁纳米簇的形成。TEM 和 HAADF-STEM 图像(图 1c-g)显示,亚纳米铁纳米簇均匀分散在 HCS 中,未形成大尺寸铁纳米颗粒。通过分析 5 个独立区域的 500 多个纳米簇以确保统计显著性和避免选择偏差(图 S3),结果一致证实纳米簇平均尺寸≈0.8 nm(图 1h)。能量色散 X 射线(EDX)mapping 验证了 Fe NCs@HCS 的元素组成(图 1e),进一步证实铁、碳、氧在整个碳壳中均匀分布,表明超小铁纳米簇的分散性良好。
PVD 策略的一个关键优势是通过简单调整乙酰丙酮铁前驱体用量,即可轻松调节 HCS 载体上的铁负载量。例如,将前驱体与 HCS 的质量比从 0.57:1 降至 0.44:1,可获得低负载量的 Fe NCs@HCS 样品(记为 s-Fe NCs@HCS),其纳米簇尺寸相同但铁含量降低(铁负载量详见后续讨论)。相反,将前驱体与 HCS 的比例从 0.57:1 显著提高至 0.69:1,得到较大尺寸的铁纳米颗粒(Fe NPs@HCS)。如图 1j,k 所示,与均匀分散的铁纳米簇不同,铁纳米颗粒在 HCS 中分散不均,尺寸分布较宽(4.8-8.3 nm),平均粒径为 6.2 nm(图 1h)。EDX mapping 进一步证实 Fe NPs@HCS 复合材料中铁的空间分布不均匀(图 1l)。
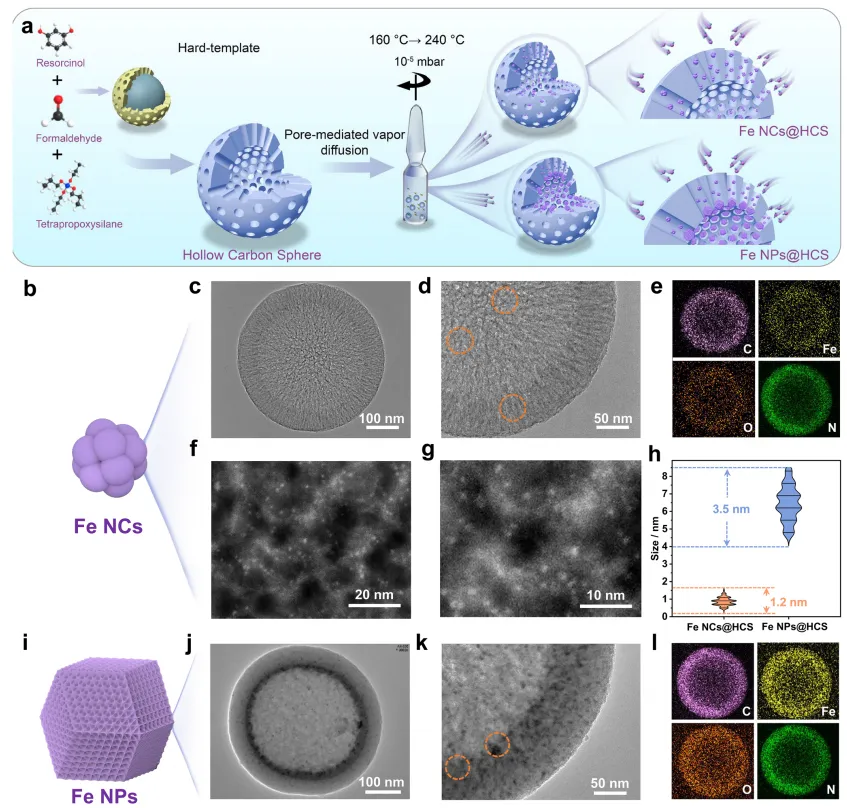
图 1 Fe NCs@HCS 和 Fe NPs@HCS 的 PVD 策略合成示意图
采用XRD分析 s-Fe NCs@HCS、Fe NCs@HCS 和 Fe NPs@HCS 的晶体结构。如图 2a 所示,与 Fe NPs@HCS 样品中观察到的明显铁衍射峰(PDF#39-1346)不同,s-Fe NCs@HCS 和 Fe NCs@HCS 的XRD图谱除在≈26° 处出现宽峰外,无其他可分辨的结晶峰,表明铁纳米簇尺寸过小,无法形成明确的晶体结构,与纳米簇的典型结构一致。通过热重分析(TGA)定量 HCS 中的铁质量负载量:400-600℃之间的重量变化对应碳的损失以及铁氧化为Fe₂O₃的过程,s-Fe NCs@HCS、Fe NCs@HCS 和 Fe NPs@HCS 中 Fe₂O₃的残留重量分别为 9.16%、12.17% 和 15.72%,对应铁含量分别为 6.41%、8.52% 和 11.00%。
孔结构在基于 PVD 的纳米簇制备策略中起着关键作用,因此通过氮气吸附 - 脱附等温线测量和孔径分布分析,对 HCS 和三种 Fe@HCS 复合材料的孔结构进行全面表征。如图 2b 所示,所有样品在高相对压力下均表现出明显的H₄型滞后环,证实介孔和微孔共存;HCS、s-Fe NCs@HCS、Fe NCs@HCS 和 Fe NPs@HCS 的比表面积分别 1152.19、1035.64、944.17 和 896.09 m²・g⁻¹。值得注意的是,铁引入后比表面积降低,且随着铁负载量增加进一步下降,表明铁物种位于 HCS 的纳米孔中。
PVD 策略下 HCS 主体中铁纳米簇与铁纳米颗粒的形成机理,与乙酰丙酮铁前驱体分子和介孔碳结构之间的负载依赖性相互作用密切相关(图 2c,d)。在此提出合理的机理:低前驱体负载量下,乙酰丙酮铁分子优先通过非共价相互作用(如氢键、范德华力)锚定在多孔通道的缺陷位点,随后在这些受限位点热解生成亚纳米铁纳米簇,此时缺陷的不饱和占据导致平均孔径适度减小(图 2d,左图);增加前驱体负载量,通道内的缺陷位点被铁纳米簇饱和占据,孔径进一步减小但未形成纳米颗粒(图 2d,中图);关键的是,前驱体过量负载时,通道缺陷完全饱和后,多余的乙酰丙酮铁分子扩散到较大的中空空腔中 —— 尽管空腔缺陷会吸附并分解形成额外的纳米簇,但非缺陷表面缺乏足够的吸附亲和力来固定剩余前驱体,这些过量的乙酰丙酮铁在热解过程中沉积到现有纳米簇表面,驱动颗粒生长为较大的铁纳米颗粒。狭窄通道内的空间限制限制了未锚定前驱体的积累,导致孔径额外减小极少;相反,广阔的中空空腔容纳了大部分多余前驱体,导致空腔壁上主要形成纳米颗粒(图 2d,右图)。TEM 成像(图 1j)显示铁纳米颗粒优先聚集在中空空腔的内壁上,证实了这一推断。此外,密度泛函理论(DFT)计算直接支持该机理:乙酰丙酮铁在缺陷碳位点上的结合能(-1.21 eV)显著强于原始通道表面(-1.11 eV;图 2c)。
为进一步验证所提出的形成机理,系统分析了 HCS 载体和 Fe@HCS 复合材料的孔径演变(图 2e;图 S8 和表 S2)。氮气物理吸附测量显示,原始 HCS 的平均孔径为 6.3 nm(图 2e);稀疏铁纳米簇沉积后(s-Fe NCs@HCS),平均直径降至 5.8 nm,这一减小与通道内部分纳米簇吸附完全一致(5.8 nm≈d₍HCS₎-d₍Fe NCs₎×1),符合低前驱体负载下缺陷限制成核的机理(图 2d,左图);饱和 Fe NCs@HCS 的直径进一步减小至 4.8 nm,与单层纳米簇形成的理论预期相符(4.8 nm≈d₍HCS₎-d₍Fe NCs₎×2),此时通道缺陷完全占据(图 2d,中图);关键的是,尽管 Fe NPs@HCS 形成了大量纳米颗粒,但孔径仍为 4.7 nm,与饱和 Fe NCs@HCS 相当,这一不变性证实多余前驱体优先迁移至中空空腔,而非进一步缩小通道,导致纳米颗粒主要在空腔壁上生长(图 2d,右图)。这种分级孔径演变为负载依赖性尺寸限制机理提供了有力证据。
采用 X 射线光电子能谱(XPS)系统研究复合材料的表面化学组成和氧化还原特性。全谱扫描结果显示铁、氧、氮、碳元素共存,证实铁纳米簇成功引入 HCS 中。高分辨率 Fe 2p 谱图(图 2f)证实三种样品中均存在 Fe (0)、Fe (II) 和 Fe (III) 物种,且随着颗粒尺寸减小,Fe (0) 的相对含量降低。这一趋势与纳米簇的典型核壳结构一致:较高氧化态(Fe (II)/Fe (III))主要来自氧化壳层,而金属 Fe (0) 来自未氧化的核部。因此,较大颗粒由于核体积相对于表面壳层更大,Fe (0) 比例更高。
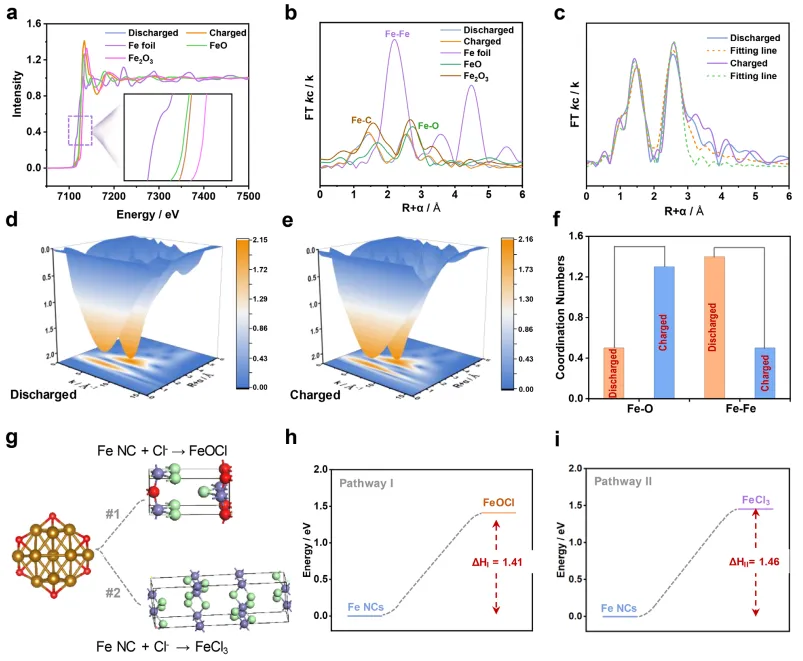
为进一步阐明电子结构和局部配位环境,进行了 Fe K 边 X 射线吸收精细结构(XAFS)光谱分析。如图 2g 所示,XANES 前边缘区域出现弱但可分辨的 1s→3d 跃迁(形式上为偶极禁戒,但因对称性破缺部分允许),表明铁中心周围存在扭曲的局部对称性。Fe NCs@HCS 的吸收边位置介于铁箔(0 价铁)和 Fe₂O₃(三价铁)之间,证实 Fe (0)、Fe (II) 和 Fe (III) 共存(图 2g)。此外,EXAFS 光谱的小波变换(WT)分析揭示了局部配位环境:5.1 和 7.4 Å⁻¹ 处的强度最大值分别对应 Fe-O 和 Fe-Fe 配位壳层(图 2h)。XPS 和 EXAFS 的联合分析表明,合成的 Fe NCs@HCS 具有混合态结构,可能为核壳构型 —— 内层核为金属 Fe (0),外层壳为 Fe (II)/Fe (III) 物种。
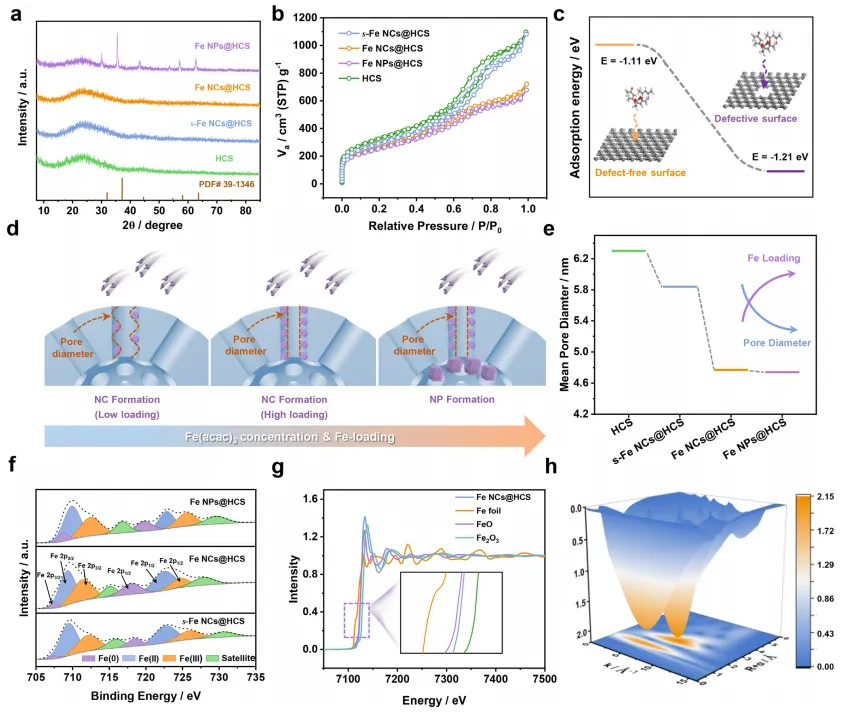
图 2 材料表征与机理洞察
电化学分析
Fe NCs@HCS表现出优于Fe NPs@HCS的电化学性能,这是高效FDI 应用的关键要求。循环伏安(CV)分析(图 3a)显示,CV 曲线呈轻微扭曲的矩形,具有 - 0.75、0.25 和 0.5 V 处的特征氧化还原峰(对应 Fe (0)/Fe (II)/Fe (III) 氧化还原反应),表现出赝电容特性。值得注意的是,Fe NCs@HCS 的最大比电容达到 480 F・g⁻¹,显著优于 s-Fe NCs@HCS 和 Fe NPs@HCS(图 3b)。这种增强的性能源于两种协同效应:首先,引入的铁纳米簇提供丰富的赝电容位点,同时保持足够的比表面积以实现快速电荷转移;其次,其超小尺寸赋予超高的表体比,促进离子扩散并最大化铁原子利用效率,共同促成优异的电化学性能。此外,尽管 s-Fe NCs@HCS 中的铁纳米簇与 Fe NCs@HCS 中的尺寸相似(图 1h),但前者的比电容仍较低(图 3b),这主要归因于 s-Fe NCs@HCS 中铁纳米簇负载量不足,无法提供足够的电荷储存氧化还原活性位点。另外,HCS 独特的球形结构增强了铁纳米簇的电荷和电子转移特性。如图 3c 的奈奎斯特图所示,高频区的半圆对应电极表面与电解质之间的电荷转移电阻(Rₙₜ):s-FeNCs@HCS、Fe NCs@HCS 和 Fe NPs@HCS 的 Rₙₜ值分别为 0.81、1.18 和 1.93 Ω,证实 Fe NCs@HCS 具有更优异的电荷转移性能。
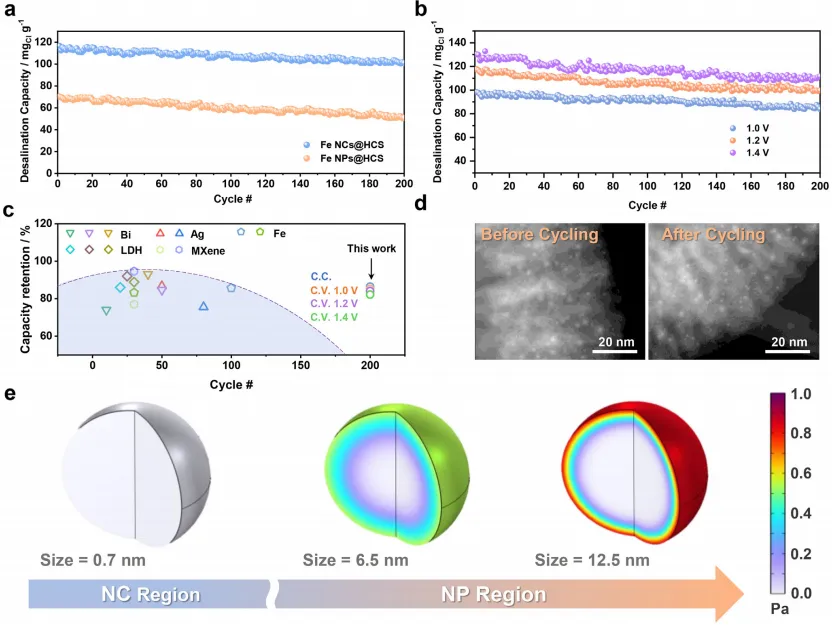
值得注意的是,Fe NCs@HCS 的 Rₙₜ显著低于 Fe NPs@HCS,这可归因于:i)电化学活性表面积增大;ii)铁纳米簇与导电碳基质之间的电子耦合更强。图 3d 展示了 Fe NCs@HCS 在不同电流密度下的恒电流充放电(GCD)曲线。所有电流密度下,GCD曲线均呈接近对称的等腰三角形,表明电荷储存行为具有高度可逆性。通过GCD测量计算的 Fe NCs@HCS 比电容均高于 Fe NPs@HCS,与 CV 结果一致。此外,Fe NCs@HCS 表现出优异的 GCD 循环稳定性,在 3.0 A・g⁻¹ 电流密度下经过 200 次循环后,电容几乎无衰减(图 3e)。
优化表面控制过程与扩散控制过程之间的平衡是同时提升离子储存容量和速率性能的有效策略,尤其优先促进表面控制过程对改善电荷储存动力学尤为有效。为定量解析这些过程对氯离子储存的贡献,在不同扫描速率下进行 CV 测试(图 S15)。如图 3f-h所示,在 10 mV・s⁻¹ 扫描速率下,s-Fe NCs@HCS、Fe NCs@HCS 和 Fe NPs@HCS 的表面贡献分别占总电容的 63.3%、62.4% 和 59.3%。这些结果表明,调节氧化还原活性材料的颗粒尺寸是调制电容行为的有效策略,而超小纳米簇可实现双模式电容,提供额外的表面控制贡献。
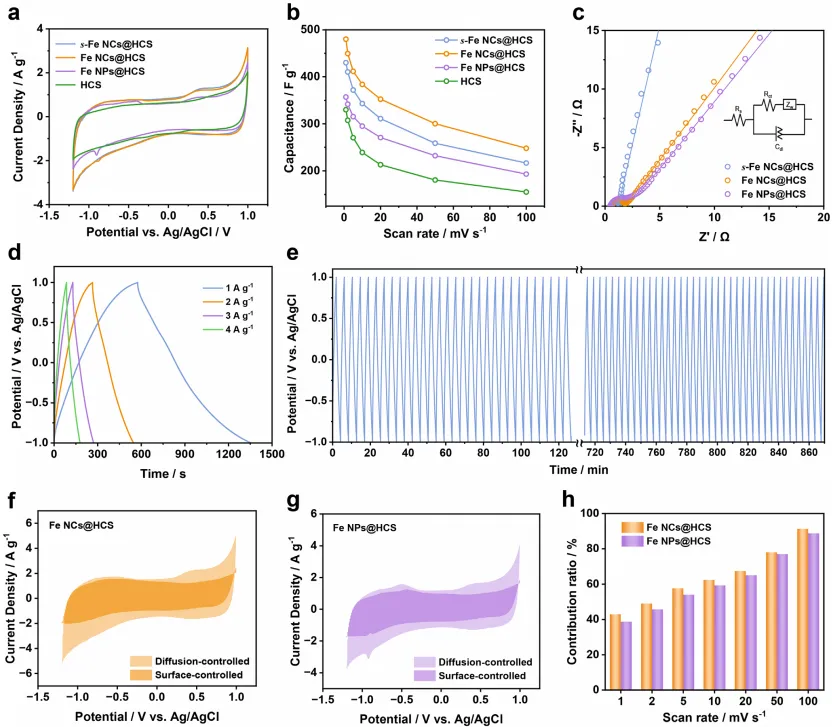
电化学反应条件
通过 Kim-Yoon Ragone 图(图 4c)进一步证实 Fe NCs@HCS 的脱盐动力学优势(数据来自不同电流密度下的脱盐测试,图 4d),该图清晰展示了其相较于 Fe NPs@HCS 更优异的脱盐容量和速率性能。与现有 FDI 体系阳极材料(银、铋、铁、层状双氢氧化物、MXene 基阳极)相比(图 4e),Fe NCs@HCS 位于性能图的最优右上区域,表明其脱盐速率极佳。这种卓越性能可能源于纳米簇结构中缩短的扩散路径,有效克服了 FDI 系统中典型的扩散限制过程。为进一步验证所提出的机理,通过有限元分析(FEA)探究尺寸依赖性扩散动力学:构建三种模型模拟不同直径的铁颗粒(0.8 nm 代表铁纳米簇、6.5 nm 代表铁纳米颗粒、12.5 nm 代表更大尺寸纳米颗粒)。模拟结果显示电解质渗透存在显著差异:对于 0.8 nm 铁纳米簇,在 0.003 s 的短时间内即可实现整个颗粒的快速均匀扩散(图4g);相反,较大尺寸纳米颗粒(6.5 和 12.5 nm)在相同时间内仅实现部分扩散,电解质渗透局限于表面区域。这种尺寸依赖性趋势表明,亚纳米簇可实现完全体相扩散,而较大纳米颗粒因电解质可及性受限,离子传输效率低下。
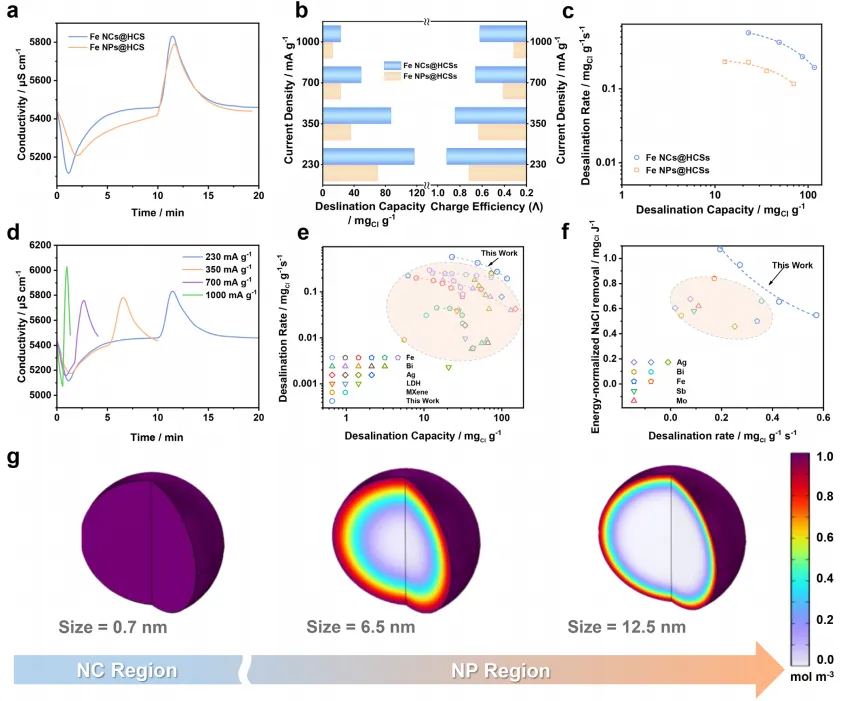
Fe NCs@HCS 基 FDI 系统相较于 Fe NPs@HCS 表现出更优异的循环稳定性,这得益于其独特的结构设计,有效解决了传统 FDI 系统中的应力相关降解问题。如图 5a 所示,在恒流运行模式下,该系统经过 200 次循环后仍保持初始脱盐容量的 86.47%,显著优于 Fe NPs@HCS 基系统(72.27% 保持率)。此外,在 1.0、1.2 和 1.4 V 下进行恒压长期测试,以更严格地评估 Fe NCs@HCS 的耐久性:如图 5b 所示,尽管与恒流模式相比容量衰减更为明显,但电极在 1.0、1.2 和 1.4 V 下仍分别保持初始容量的 85.2%、84.1% 和 82.2%。同时,在宽盐度范围(100-35000 mg・L⁻¹)内进行多循环脱盐测试:低盐浓度下初始脱盐容量近乎线性增加,盐度超过≈1000mg・L⁻¹后最终稳定在≈110 mg₍Cl₎・g⁻¹,这是电容去离子(CDI)和吸附系统的典型趋势,进一步证明该系统对不同盐度的适应性。此外,测试结果显示初始盐度与容量保持率呈负相关,且在≈1000 mg・L⁻¹ 时趋于稳定:稀电解质中,由于离子匮乏,活性材料初始利用率较低,长期循环过程中容量可逐步恢复;而高盐度下,充足的离子供应确保初始循环中氯离子储存位点快速完全占据,导致容量早期饱和,进而保持率稳定。此外,将Fe NCs@HCS 基 FDI 系统的循环性能与先前报道的铁基 FDI 系统进行系统对比(图 5c),结果显示 Fe NCs@HCS 基 FDI 在保持更高脱盐容量的同时,循环寿命显著延长,凸显其卓越的长期稳定性。
这种增强的稳定性源于两种协同机理:i)共形碳壳提供物理限制,抑制铁纳米簇聚集和溶解;ii)超小纳米簇尺寸(0.8 nm)从根本上减少循环过程中的应力积累,避免碳壳开裂和变形。为验证这些假设,进行了全面的循环后形貌表征和 FEA 分析:TEM 和 FESEM 观察显示,Fe NCs@HCS 经过 200 次循环后仍保持显著的结构稳定性,铁纳米簇维持原始尺寸,中空碳球保留球形形貌和多孔结构(图 5d);相比之下,Fe NPs@HCS 表现出显著的结构降解,多次循环过程中的应力积累导致碳壳出现大量裂纹和断裂(图)。除形貌表征外,通过电感耦合等离子体(ICP)分析确定铁纳米簇和铁纳米颗粒电极的铁溶出程度:结果显示,铁纳米簇电极的铁溶出量可忽略不计(0.57 μg・L⁻¹),而铁纳米颗粒电极的铁溶出量为 8.4 μg・L⁻¹,形成鲜明对比。FEA 为这些观察结果提供了定量支持(图 5e):模拟结果表明,将活性材料尺寸从纳米颗粒减小至 0.8 nm 纳米簇,最大内应力降低≈99.41%,有效防止碳壳断裂。这种尺寸依赖性应力降低机理解释了 Fe NCs@HCS 基系统优异的循环稳定性,为克服 FDI 中应力诱导性能降解的长期挑战提供了关键见解,并证实活性材料的亚纳米工程可同时解决电化学脱盐系统中的动力学和稳定性问题。
机理研究
为解决这一机理争议,采用基于同步辐射的 Fe 傅里叶变换扩展 X 射线吸收精细结构(FT-EXAFS)分析和 Cl X 射线吸收近边结构(XANES)分析 —— 该技术的高 k/R 空间分辨率能够精确追踪 Fe 配位环境。氯化过程中(图 6a-e),观察到 Fe-O 配位数增加而 Fe-Fe 配位数降低(图 6f)。这种结构演变唯一支持路径 I(Fe→FeOCl),因为 FeOCl 的形成需要增强 Fe-O 键合,同时破坏金属 Fe-Fe 键。DFT 计算进一步证实了该路径:FeOCl 形成的反应焓(ΔH I=1.41 eV)低于 FeCl₃(ΔH II=1.46 eV)(图 6g-i)。此外,脱盐过程后 Fe NCs@HCS 的 Cl K 边 XANES 测量也证实了 FeOCl 的形成。EXAFS/DFT 的联合证据明确表明,Fe NCs@HCS 中的氯离子储存通过转化反应生成 FeOCl 进行,为铁基 FDI 阳极的发展提供了原子级的机理清晰度。
总结



