青岛科技大学AEM:具有增强界面溢出效应的Os-OsP2的超快微波准固态构建用于海水基阴离子交换膜电解槽
- 2026-05-28 19:44:37

1、文章亮点
报道了一种通过超快(20 s)微波准固相方法制备的具有异质结的锇-磷化锇(Os-OsP2)催化剂,Os原子和OsP2基团在提高催化效率方面起到了关键作用,缓慢的水解解离相主要是由Os加速的,而随后的氢吸附过程主要是由OsP2促进。
碱性交换膜水电解槽时,碱性海水,Os-OsP2 1.74V @ƞ1000。稳定性100h@ƞ1000。
实验和理论分析表明,Os-OsP2界面优化了电子结构:锇(Os)位点通过降低d带中心加速水的解离,而OsP2通过界面溢出促进氢的脱附,共同降低了HER的能垒。
2、全文速览
开发具有成本效益的析氢反应(HER)催化剂以替代碱性海水介质中的Pt/C仍然是一项关键挑战。因此,本文报道了一种通过超快(20 s)微波准固相方法制备的具有异质结的锇-磷化锇(Os-OsP2)催化剂,用于工业级电流密度下的海水分解。实验和理论分析表明,Os-OsP2界面优化了电子结构:锇(Os)位点通过降低d带中心加速水的解离,而OsP2通过界面溢出促进氢的脱附,共同降低了HER的能垒。此外,该催化剂在阴离子交换膜水电解槽中仅需1.74 V即可达到1 A cm-2,且具有较高的价格活性,在相同条件下效率比商业Pt/C高出23%。进一步研究表明,它在宽pH范围内均表现出优异的HER活性,并且在碱性海水中具有超过100 h的卓越耐久性。经济评估显示其具有优异的成本活性(85.6 A dollar-1),是Pt/C的90倍,制氢成本($0.86 GGE-1)低于美国能源部(DOE)的目标。本研究为开发用于海水规模化制氢的高性能、低成本催化剂提供了可行的指导。
3、背景介绍
氢被视为一种环境友好型能源载体,有望实现碳中和。通过电催化水分解可高效制氢,该过程还能将电能转化为化学能。海水资源丰富且易于获取,已成为淡水电解的理想替代方案,有效缓解了淡水资源短缺的问题。在各种电解液中,碱性海水电解液具有独特的优势。与酸性环境相比,碱性溶液提供了更宽的电势窗口(高达480 mV),显著降低了氯氧化反应(ClOR)与析氧反应(OER)竞争的风险。此外,在海水电解中,阴极的析氢反应(HER)对制氢效率起着关键作用。然而,整体电解速率主要受阳极反应的选择性和电解槽的耐腐蚀性限制。为提高海水电解的综合性能,必须协同优化HER催化剂和阳极材料。因此,深入研究海水电解及其与能源装置的集成应用,尤其是在工业环境中,对于增强能源可持续性和优化其在氢经济中的作用至关重要。
HER催化剂的本征活性主要由氢吸附自由能(ΔGH*)和水离解能垒决定。铂族金属(PGMs)及其化合物通常表现出最优的ΔGH*值,位于HER火山图的顶端。其中,铂(Pt)因其在酸性介质中反应动力学快且氢键强度高,成为商业上公认的HER催化剂。然而,在非酸性介质中,水离解能垒较高,加之成本高和长期稳定性问题,限制了其广泛应用。其他铂族金属如铑(Rh)、钯(Pd)和铱(Ir)比Pt更昂贵,无法作为替代催化剂。钌(Ru)成本相对较低,且催化性能与Pt相似,是一种很有前景的替代品,并已用于HER研究,如Ru/RuS2、Ru-Ru2P和Ru SAs@PN。同时,锇(Os)作为所有铂族金属中价格最低的元素,具有与Ru相似的d带结构。开发Os基催化剂可作为贵金属催化剂体系的技术储备。通过将Os与非贵金属耦合以减少Os用量的技术进步,能有效缓解未来成本上涨问题,并拓宽Os基催化剂的应用前景。然而,关于Os基纳米材料用于电催化应用的报道较少。这一现象主要归因于Os基材料的强吸附特性,Os位点的解吸能垒显著较高。因此,必须采用适当的方法调节Os基纳米材料的电子态及其后续吸附行为,以最大限度地提高催化活性。
在此,本文成功合成了具有显著增强碱性海水中析氢反应(HER)性能的锇-磷化锇(Os-OsP2)复合材料,并很好地阐明了其潜在的促进机制。通过将实验结果与密度泛函理论(DFT)计算相结合,本文发现Os结构单元能有效促进水分解生成H*,然后通过Os-OsP2界面的溢流效应将H*高效转移到OsP2位点。这种转移增加了该位点上的H*覆盖率,从而提高了HER效率。Os-OsP2异质界面的存在进一步提升了产氢效率。值得注意的是,当在阴离子交换膜水电解槽(AEMWE)中与NiFe泡沫集成时,Os-OsP2复合材料在海水中仅需1.74 V即可达到1 A cm-2的高电流密度,实现了每加仑汽油当量(GGE)下高效且低成本的产氢。该性能显著优于Pt/C基对应物。这些发现激发了本文在用于实际产氢的贵金属基电催化剂中实现成本效益与高活性之间平衡的兴趣。
4、图文解析
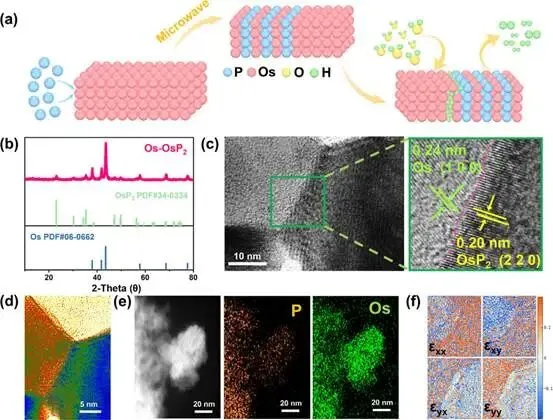
图1. a) Os-OsP2合成路径的示意图。b) Os-OsP2的X射线衍射图。c) Os-OsP2的高分辨率透射电镜图像。d) Os-OsP2的强度映射图。e) Os-OsP2各元素的能量色散X射线光谱。f)对应(100)和(220)晶面的晶格应变exx、exy、eyx和eyy。
图1a展示了催化剂Os-OsP2的合成路径以及制氢过程的示意图。所设计催化剂中的Os组分对水表现出强亲和力,而OsP2则具有优异的H*脱附性能,可有效促进氢气的产生。为验证合成催化剂的晶体结构,进行了X射线衍射(XRD)分析(图1b)。通过准固态微波辅助燃烧法合成的产物主要由两相组成:OsP2(PDF#340334)和Os(PDF#06-0662)。这两相的协同组合赋予了电催化性能独特的优势。扫描电子显微镜(SEM)图像显示其为致密的块状结构,这确保了材料的结构稳定性和均匀的电荷分布。整体构型有利于构建稳健的电子传输路径,同时保持足够的电化学活性位点。采用透射电子显微镜(TEM)进一步观察Os-OsP2样品的微观形貌。整体形貌呈现出纳米薄片和颗粒结构的混合体。放大后(图1c),Os和OsP2之间的异质结界面清晰可见,其中0.24 nm的间距对应于Os的(100)晶面,0.20 nm的间距属于OsP2的(220)晶面。OsP2的(220)晶面。[18] TEM图像的强度映射分析也揭示了明显的异质结合现象(图1d)。界面结构对于催化反应过程中的电子转移和物质传输至关重要。能量色散X射线光谱(EDS)元素分布图(图1e)证实了Os和P在整个样品中均匀分布。几何相位分析(GPA)算法从TEM图像中提取应变分布。如图1f所示,在异质结界面处观察到显著的应变差异,随着远离该界面,应变值逐渐趋于平稳。此外,一些晶格条纹显示出波浪形畸变或原子排列的局部无序化,这可能是由于应变效应引起的。这些应变效应有利于暴露更多的活性位点并促进催化反应。
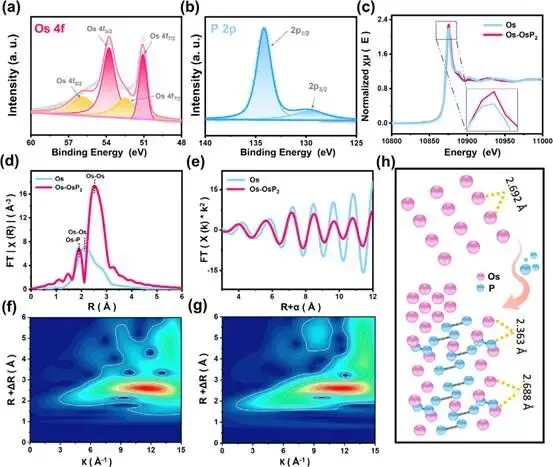
图2. Os-OsP2的XPS谱图 a) Os4f。b) P2p。c) Os L3边XANES以及d) Os箔和Os-OsP2相应的EXAFS谱图。e) Os-OsP2中的Os L3边EXAFS振荡。f) Os和g) Os-OsP2的EXAFS信号的WT。h) Os和Os-OsP2的模型示意图。
X射线光电子能谱(XPS)分析表明,Os、OsP2和Os-OsP2的成分均包含Os和P。对于Os-OsP2中的Os4f,Os4f5/2(53.69 eV)和Os4f7/2(51.08 eV)的峰归属于Os4+,Os4f5/2(55.69 eV)和Os4f7/2(52.93 eV)归属于Os0,这表明Os-OsP2中这两种价态共存,且结合能相对于纯Os和OsP2向更高方向偏移了≈0.2 eV(图2a)。如图2b所示,对应于P2p1/2(130.57 eV)、P2p3/2(129.66 eV)和PO(134 eV)的峰,在Os-OsP₂中与纯OsP2相比偏移了≈0.3 eV。这种现象可能归因于Os和OsP2之间的电子转移,Os─P键促进了这种转移。P2p和Os4f的组合光谱进一步证实了OsP2和Os-OsP2中Os-P键的形成。这也证明了界面处Os和OsP2之间的电子相互作用。为了进一步确定Os原子的价态和局部配位环境,对纯Os和异质结构Os-OsP2的Os L3边进行了X射线吸收近边结构(XANES)和扩展X射线吸收精细结构(EXAFS)分析。如图2c所示,Os-OsP2在Os L3边XANES中的白线峰强度和吸收边位置与Os箔无限接近。此外,在一阶导数XANES曲线中,Os-OsP2的E₀峰非常接近Os箔。这些结果表明,异质结构Os-OsP2的形成使Os物种的平均价态接近0价。此外,Os L3边的k³χ(k)振荡光谱和未经校正的相位校正EXAFS傅里叶变换结果(图2d)显示,Os-OsP2的信号特征更接近Os箔。相应的EXAFS拟合结果显示,每个Os原子与≈6.0个Os原子和2.0个P原子配位,Os-Os和Os-P的距离分别为2.363和2.688 Å(图2e)。值得注意的是,与Os相比,Os-OsP2中Os-Os和Os-P的配位数(CN)和距离(R)发生了变化(CNOs-Os=6,ROs-Os=2.692 Å,),这进一步表明异质界面的形成诱导了Os与Os以及P之间的原子相互作用,导致Os-Os和Os-P键的形成,这与电子局域函数(ELF)结果一致(图5c)。最后,小波变换(WT)-EXAFS(图2f、g)进一步可视化了Os-OsP₂异质结构中Os-Os和Os-P路径的共存。上述光谱分析剖析了Os-OsP2的异质电子结构(图2h)。
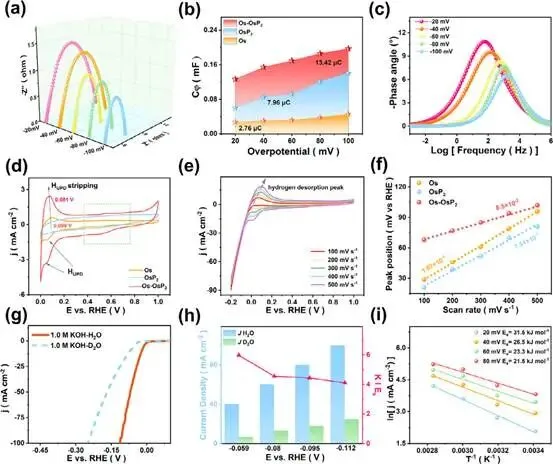
图3. 在1.0 mol/L KOH中 a) Os-OsP2催化剂在不同电位下的奈奎斯特图。b) HER过程中催化剂的Cφ随电位变化图。c) Os-OsP2的波特图。d)50 mV/s下的循环伏安曲线。e)氩气氛围中不同扫描速率下记录的循环伏安曲线。f)催化剂的氢脱附峰位置随扫描速率变化图。g) Os-OsP2催化剂在1 mol/L KOH-H2O和1 mol/L KOH-D2O溶液中的极化曲线。h)动力学同位素效应值随电位变化图。i) Os-OsP2的阿伦尼乌斯图。
为了研究HER中的氢吸附/解吸过程,进行了原位EIS测试以验证H在Os-OsP2上的吸附强度。通过等效电路模型对奈奎斯特图进行了模拟(图3a)。此外,Cφ也可以通过之前使用的等效电路模型计算得出,其表示与过电位相关的吸附H物种覆盖率的表观赝电容。氢吸附电荷(QH)是用于描述HER过程中催化剂表面H量的值,通过对Cφ积分进行定量计算。与Os和OsP2相比,Os-OsP2的QH显著增加(图3b),表明引入的P和生成的界面可以促进水分解为H*。此外,基于EIS的塔菲尔斜率与过电位之间的关系可以量化Os-OsP2的氢吸附动力学,结果表明Os-OsP2的氢吸附动力学更快,能够为HER提供充足的质子供应。类似地,从波特图获得的相位峰角随电压的增加而减小(图3c),这表明电荷转移阻抗降低。在相同电位下,Os-OsP₂的相位峰角低于Os和OsP2,表明Os-OsP2具有更快的HER动力学。在图3d中,Os-OsP2比Os表现出更明显的氢欠电位沉积(HUPD)峰,表明在HER过程中更多的H*吸附在Os-OsP2表面。HBe与HUPD(E峰)相关,表明Os-OsP2的氢结合能(Hbe)值(0.081 V)低于Os(0.099 V),表明Os-OsP2的氢脱附强度相对OOS较低(图3e)。换句话说,在良好的吸水条件下,Os-OsP2表面更多的活性位吸附了H,这反过来又有利于HER。为了说明氢的脱附过程,在图3e中,测试了不同扫描速率下的循环伏安(CV)曲线,图3e中可以观察到明显的氢脱附峰,并且随着扫描速度的增加,峰的位置向高电压移动。因此,峰的位置与扫描速度进行了拟合,以评估氢气的脱附动力学。在图3f中,Os-OsP2的斜率显著下降,表明H*解吸动力学加速。此外,OH*的吸附对碱性HER也是必不可少的。图3d的局部放大图,其中可以观察到清晰的Os-OsP2的OH*吸附/脱附峰。较好的OH*吸附/脱附能力有利于活性中心的释放。通过动力学同位素实验(KIE)深入研究了H*的动力学。图3g显示了Os-OsP2在1.0mKOH-H2O和1.0mKOH-D2O溶液中的LSV曲线。相应的KIE值,由H2O和D2O的电流密度比(JH2O/JD2O)得出,如图3h所示。对于Os-OsP2,达到40、60、80和100mA cm-2的电流密度所需的电位分别为−0.059、−0.08、−0.095和−0.112 V(与RHE相比)。相反,在相同电位下,在1.0mKOH-D2O溶液中测得的Os-OsP2的电流密度明显较低。计算了每个势的KIE值,表明氢转移是限速过程中的关键步骤。为了探索异质结界面对反应动力学活化能的影响,收集了不同温度下Os-OsP2、OsP2和Os的催化活性。图3i说明了催化活性随着温度的升高而增强。使用Arrhenius方程(公式2)计算了不同温度下20、40、60和80 mV过电位下的电流密度。结果表明,在相同的电势下,Os-OsP2的活化能低于Os和OsP2的活化能,表明异质结界面增强了HER的动力学。
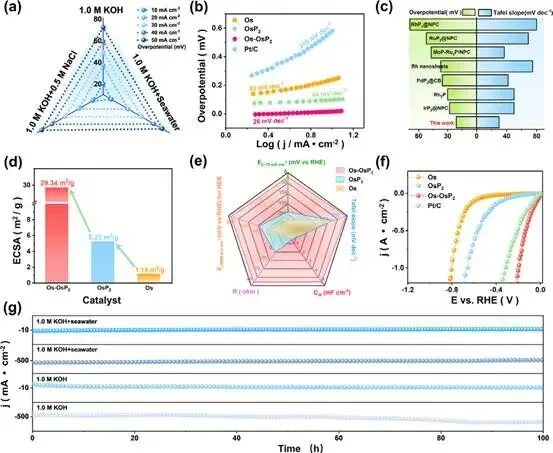
图4 a)Os-OsP2在1.0mKOH+海水、1.0mKOH、1.0mKOH+0.5mNaC l中的LSV曲线。b)催化剂在1.0m KOH中的塔菲尔斜率。c)将Os-OsP2催化剂的性能与最近报道的催化剂进行了比较。d)催化剂的ECSA。(e)Os、OsP2和Os-OsP2在1.0mKOH+海水中的电化学性能。f)高电流密度下的LSV曲线。g)OS-OsP2的稳定性试验。
用常规的三电极体系评价了Os-OsP2在碱性电解液中对HER的电化学性能。结果表明,当电流密度为10 mA cm-2时,在1.0mKOH、1.0mKOH+海水和1.0mKOH+0.5mNaCl溶液中的过电位分别为20 mV、20 mV和24 mV。这些较低的过电位表明,Os-OsP2在碱性介质中具有良好的催化性能。此外,与Os和OsP2相比,Os-OsP2的塔菲尔斜率显著降低到26 mV(图4b),表明其速率控制步骤从Os的水解离转变为OsP2的氢复合。Os-OsP2在这些方面的表现优于大多数已报道的贵金属基催化剂材料(图4c)。电化学阻抗谱(EIS)表明,Os-OsP2具有比Os和Os-OsP2更低的电荷转移电阻,这表明异质结界面增强了电子转移和HER动力学。从不同扫描速度下的循环伏安曲线计算了双电层电容(CDL)和电化学活性表面积(ECSA)(图4d)。Os-OsP2的ECSA越大,活性中心越多,催化性能越好(方程式1)。值得注意的是,OsP2的性能没有受到影响,显示了它的稳健性和对不同水环境的适应性。此外,对10和500 mA cm-2电流密度下的Tafel斜率、CDL、EIS和过电位进行了综合分析,结果如图4e所示,其中Os-OsP2的电催化性能明显优于其他参考样品。为了测试催化剂在工业应用中的性能,在高电流密度下进行了线性扫描伏安法测试,Os-OsP2(在1A cm-2时为192 mV)的性能也优于Pt/C(图4f)。用I-t法对催化剂的稳定性进行了评价,结果表明,催化剂的电流密度在100h内几乎没有衰减,表明其在碱性和天然海水中都具有良好的稳定性(图4g)。尽管在碱性条件下的研究已经取得了重大进展,特别是在常规的碱性电解液体系和碱性海水环境中,但现实世界应用场景的复杂性对催化剂的性能提出了更高的要求。
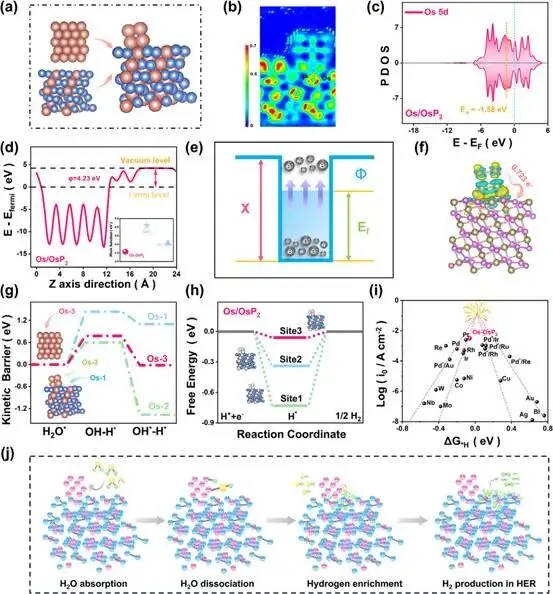
图5.a)Os、OsP2和Os-OsP2的结构模型。b)Os-OsP2的ELF图(红区表示电荷聚集,蓝区表示电荷耗散)。c)计算了Os-OsP2模型的表面或界面Os位置的d带中心。d)Os-OsP2的功函数。e)电子转移示意图。f)Os-OsP2的DCD。g)水在不同部位的解离动力学势垒。h)不同位置上的氢析出。i)HER的Os-OsP2(红色正方形)和常见金属催化剂(黑色圆圈火山图))。j)Os-OsP2的碱水分解示意图。
通过密度泛函理论(DFT)模拟,阐明了Os-OsP2异质结构对HER的催化作用,并准确地确定了吸附/解离条件和氢吸附势垒。本文已经成功地构建了异质结构的Os-OsP2模型,清楚地显示了纯相和非均相界面(图5a)。从ELF图(图5b)可以观察到,OsP2在Os和OsP2之间显示了较少的电荷局域化,有效地避免了H+在两相界面上的积累。综上所述,可以看出,吸附的氢与非均相界面上的活性中心之间的相互作用明显减弱,导致脱附能垒显著降低。当进一步探索Os和Os-OsP2系统时,发现电子结构上的显著差异,这些差异对HER的表现有深刻的影响。具体地说,Os的d带中心在-1.8 eV,而Os-OsP2的d带中心在−1.58 eV,这清楚地表明Os-OsP2的d带中心上移到费米能级(图5c)。d带中心上移加强了Os的d轨道和H反键轨道之间的重叠,优化了吸附-脱附平衡。这意味着OsP2体系中Os原子的d轨道电子态更加接近费米能级,从而大大增强了它们与吸附物的相互作用。当Os和OsP2结合形成Os-OsP2时,功函数进一步下降到4.23 eV,强烈表明界面效应或结构优化策略可以显著增强材料的电子传输能力,使电子更容易从电极表面释放并积极参与HER反应(图5d,e)。为了更深入地了解HER过程中电子结构的变化,进行了微分电荷密度(DCD)分析(图5f)。计算结果表明,当氢中间体吸附在催化剂表面时,DCD修饰中心周围的电荷密度发生了显著的变化。这种电子的重新分布改善了催化剂与氢中间体之间的相互作用,促进了氢在HER机理中的吸附和脱附步骤。与Os和OsP2相比,Os-OsP2的水解离能垒成功地降低到0.78 eV(图5g),证实了异相界面的构建可以有效地促进非酸性介质中吸附氢的形成,从而显著加快产氢速度。在HER的研究中,氢吸附的吉布斯自由能(ΔGH*)被认为是其性能的核心指标(图5h)。纯相Os和OsP2的ΔGH*值较低,分别为−0.34 eV和−0.73 eV,表明在Os和OsP2表面的活性位上氢的脱附动力学受到很大阻碍。当多相界面构建成功后,Os-OsP2模型界面吸附位置的ΔGH*值优化为−0.06 eV,这意味着H*更倾向于在OsP2上转化生成氢气。已经精确地确定了Os-OsP2模型在HER火山图中的确切位置(图5i)。在HER的研究领域,原位拉曼光谱已经成为探索Os-H键复杂特征的有力工具。在0V时,Os-─氢键出现了明显的特征,其强度起伏是金属-氢相互作用动态性质的实时指示器。在萌芽阶段,这一特征峰微弱而宽广,表明Os-H键处于萌芽、不稳定状态。随着外加电压的增加,与Os-H键相关的拉曼信号的强度随之增加,伴随着峰轮廓的锐化。这一演变意味着金属和氢之间相互作用的加强,导致氢原子在金属表面上的稳定和吸附。这些洞察力对于为氢气生产的后续阶段奠定基础至关重要。因此,这种方法能够仔细地揭示高温条件下HER过程背后的微观动力学。详细的机理如图5j所示。H2O在Os附近吸附并解离生成H*,然后H*被吸附到OsP2上生成H2。总而言之,Os-OsP2异质界面在全pH介质中表现出良好的电催化HER动力学。
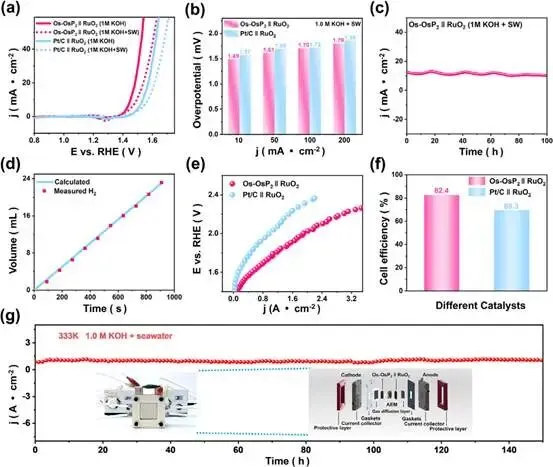
图6.a)Os-OsP2‖RuO2和Pt/C‖RuO2的整体水分裂的LSV曲线。b)比较在碱性海水中不同电流密度下所需的电压。c)Os-OsP2‖RuO2在1.0mKOH+海水中的计时电流曲线。d)产气量随时间变化的理论值和实际值。e)AEM电解槽中总的海水电解LSV曲线。f)比较了Os-OsP2‖RuO2和Pt/C‖RuO2的催化效率。(f)1A cm-2处的计时安培曲线和AEM电槽的照片。
5、总结与展望
综上所述,本工作精心设计了一种既具有高活性又具有高耐久性的OS-OsP2电催化剂。通过大量的电化学测试、原位分析技术和密度泛函计算,本文证实了所有的Os原子和OsP2基团在提高催化效率方面起到了关键作用。值得注意的是,缓慢的水解解离相主要是由Os加速的,而随后的氢吸附过程主要是由OsP2促进的。这种协同效应显著地提高了催化剂在1mKOH下的性能。值得注意的是,Os-OsP2在工业环境中表现出出色的稳定性,在100h的不间断操作中保持不变的电流密度。经济评估显示,将该催化剂集成到Os-OsP2‖RuO2配置中,在500 mA cm-2时,太阳能到氢的转换效率达到了令人印象深刻的71.6%,超过了传统Pt/C‖RuO2配置的65.7%基准。此外,使用OsP2生产氢气的成本远远低于美国能源部设定的门槛,这突显了其非凡的成本效益。因此,Os-OsP2不仅是一种高效、经济的催化剂,而且在可持续制氢的大规模海水电解应用中也是一个很有前途的候选者。
6、文章链接
Ultrafast Microwave Quasi-Solid-State Construction of Os-OsP2 with Enhanced Interfacial Spillover for Seawater-Based Anion Exchange Membrane Electrolyzers
https://doi.org/10.1002/aenm.202501054


如需转载或投稿请联系我们:energy_catalysis@163.com
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 青岛海鲜市场犹如“春运现场”,一斤大虾28元,当地人成箱购买
- 青岛司法丨致全市监狱、戒毒人民警察及家属们的慰问信
- 熊叭竞技俱乐部,限青岛地区2档及以下《2026年新手大奖赛》将于1月12日举办.
- 欢度春节-2月出境旅行清单-青岛起止
- 欢度春节-2月国内旅行清单-青岛起止
- 豪诚台球俱乐部,限青岛地区3档及以下《中式八球挑战赛》将于1月11日举行.
- 医保 | 青岛市长期护理保险“居家照护”服务指南
- 年均接处警4.85万起!青岛“最忙”派出所的法治密码
- 青岛平度:“美丽经济”绽放 蝴蝶兰“飞”向世界
- 青岛、济南、威海等地招聘厨师2万起/东北菜1万/湘菜炒锅7K/家常菜8.5K
